Category: STEM (Science, Technology, Engineering and Mathematics)
ORIGINAL
An Upgraded Blended Model for Human Protein Classification Using Fast Spec CNN with Multi-Head Attention and GAN Augmentation
Un modelo mixto mejorado para la clasificación de proteínas humanas utilizando una CNN Spec rápida con atención multicabezal y aumento de GAN
Savitha S1 ![]() *, Kalai Vani Y S1
*, Kalai Vani Y S1 ![]() *, Umme Najma2
*, Umme Najma2 ![]() *, Komala K V3
*, Komala K V3 ![]() *, Deepa V P4
*, Deepa V P4 ![]() *, Jyothi N M1
*, Jyothi N M1 ![]() *
*
1UBMS Institute of Technology and Management, Information Science and Engineering. Bengaluru, India .
2Government Science College, Department of Biotechnology. Hassan, India.
3Government Engineering College, Computer Science and Engineering. Ramanagara, India.
4Government Engineering College, Department of Electronics and Communication. Ramanagara, India.
5Koneru Lakshmaiah Education Foundation, Computer Science Engineering. Vaddeswaram, India.
Cite as: Savitha S, Vani Y S K, Najma U, K V K, V P D, N M J. An Upgraded Blended Model for Human Protein Classification Using Fast Spec CNN with Multi-Head Attention and GAN Augmentation. Salud, Ciencia y Tecnología - Serie de Conferencias. 2024; 3:804. https://doi.org/10.56294/sctconf2024804
Submitted: 10-01-2024 Revised: 12-04-2024 Accepted: 14-07-2024 Published: 15-07-2024
Editor:
Dr.
William Castillo-González ![]()
ABSTRACT
Introduction: proteins play a critical role in cellular functions, and the evaluation of protein patterns in microscope images is vital for biomedical research. This study introduces a pioneering hybrid framework for human protein classification, leveraging a combination of Fast Spectral Convolutional Neural Network (CNN) with Multi-Head Attention and GAN Augmentation. This innovative approach aims to mechanize the examination of microscope images containing mixed protein patterns, thereby accelerating biomedical research insights into human cells and diseases.
Method: the framework integrates spectral processing layers and attention mechanisms into the Fast Spec CNN architecture to enhance classification accuracy and interpretability. Through GAN augmentation, synthetic protein images are generated to complement the real dataset, bolstering model generalization and robustness. The Fast Spec CNN model, coupled with Multi-Head Attention, adeptly captures spectral features and discerns discriminative representations.
Results: the study achieved an impressive accuracy rate of 98,79 % on the Image segmentation of the Human Protein Atlas dataset, outperforming prior methodologies. The results underscore the efficacy of the suggested model in accurately classifying proteins across various hierarchical levels simultaneously. GAN augmentation enriches dataset variability and fortifies model resilience.
Conclusions: this study makes significant additions to automated biomedical image analysis, providing a valuable tool for the expedited exploration of human cells and diseases. The architectural flexibility of the emulate enables end-to-end processing of protein images, offering interpretable representations and profound insights into cellular structures and functions. Compared to earlier studies, such as UNet, DeepHiFam with ProtCNN, ProPythia, Protein Bert, ELM, and CNN, this framework performs better than others in terms of accuracy, achieving 98,79 %, the highest among the compared methodologies.
Keywords: Human Protein Classification; Fast Spectral CNN; Multi-Head Attention; GAN Augmentation; Biomedical Image Analysis.
RESUMEN
Introducción: las proteínas desempeñan un papel crítico en las funciones celulares, y la evaluación de los patrones de proteínas en imágenes de microscopio es vital para la investigación biomédica.
Este estudio introduce un marco híbrido pionero para la clasificación de proteínas humanas, aprovechando una combinación de redes neuronales convolucionales espectrales rápidas (CNN) con atención multicabezal y aumento de GAN. Este enfoque innovador tiene como objetivo mecanizar el examen de imágenes de microscopio que contienen patrones mixtos de proteínas, acelerando así la investigación biomédica en células humanas y enfermedades.
Método: el marco integra capas de procesamiento espectral y mecanismos de atención en la arquitectura Fast Spec CNN para mejorar la precisión de la clasificación y la interpretabilidad. Mediante el aumento de GAN, se generan imágenes sintéticas de proteínas para complementar el conjunto de datos reales, reforzando la generalización y robustez del modelo. El modelo Fast Spec CNN, junto con Multi-Head Attention, captura hábilmente las características espectrales y discierne las representaciones discriminatorias.
Resultados: el estudio alcanzó una impresionante tasa de precisión del 98,79 % en la segmentación de imágenes del conjunto de datos del Atlas de Proteínas Humanas, superando a metodologías anteriores. Los resultados subrayan la eficacia del modelo propuesto para clasificar con precisión proteínas en varios niveles jerárquicos simultáneamente. El aumento de GAN enriquece la variabilidad del conjunto de datos y fortalece la resistencia del modelo.
Conclusiones: este estudio aporta importantes adiciones al análisis automatizado de imágenes biomédicas, proporcionando una valiosa herramienta para la exploración acelerada de células y enfermedades humanas. La flexibilidad arquitectónica del emulador permite el procesamiento integral de imágenes de proteínas, ofreciendo representaciones interpretables y profundos conocimientos de las estructuras y funciones celulares. Comparado con estudios anteriores, como UNet, DeepHiFam con ProtCNN, ProPythia, Protein Bert, ELM, y CNN, este marco obtiene mejores resultados que los demás en términos de precisión, alcanzando un 98,79 %, el más alto entre las metodologías comparadas.
Palabras clave: Clasificación de Proteínas Humanas; CNN Espectral Rápida; Atención Multicabezal; Aumento de GAN; Análisis de Imágenes Biomédicas.
INTRODUCTION
Proteins play a critical role in cellular functions, and the assessment of protein patterns in microscope images is vital for biomedical research. High-throughput microscopy generates these images at a pace exceeding manual evaluation capabilities, necessitating automated image analysis for accelerated understanding of human cells and diseases. This study focuses on classifying mixed protein patterns in microscope images, a multilabel problem where images can belong to multiple classes. The class labels represent various cellular components, including mitochondria, nuclear bodies, nucleoli, Golgi apparatus, nucleoplasm, nucleoli fibrillar center, cytosol, plasma membrane, centrosome, and nuclear speckles. The principal aim of this probe is to develop an advanced blended model for human protein classification by integrating Fast Spectrogram CNN (Spec CNN), a Multi-Head Attention mechanism, and GAN-based data augmentation. This innovative approach aims to enhance automated biomedical image analysis, thereby facilitating efficient exploration of protein patterns in cells.
The inquiry is required due to the limitations of current methodologies in achieving high clarity along with efficacy in protein classification. Existing techniques often fall short in handling the complexity and diversity of protein patterns. By pioneering the exploitation of Fast Spectrogram CNN with Multi-Head Attention and GAN-based data augmentation, this research seeks to plug a sizable gap in the industry. Experimental results demonstrate our model’s exceptional accuracy of 98,79 % on the Visual Interpretation of the Human Protein Atlas. This remarkable result underscores the effectiveness of our hybrid framework in accurately classifying mixed protein patterns in microscope images. This research significantly contributes to automated biomedical image analysis, providing a valuable tool for the expedited exploration of human cells and diseases. The advancements presented in this study have the potential to significantly impact medicine and biology, offering a more efficient and accurate means of analyzing protein patterns in biomedical research. Through the integration of deep learning and innovative techniques, we propel advancements in medicine and biology.
The literature survey conducted is highlighted. The training and validation of the DPN U-Net neural network utilized a dataset comprising 266 images sourced from the Human Protein Atlas. Training the model to convergence was accomplished using a single Nvidia GeForce RTX 2070 graphics card, which took approximately 1,5 hours.(1) Sc Linear is a versatile tool designed to predict single-cell protein levels from gene expression using a linear regression approach. Developed as an end-to-end framework, it encompasses all essential steps of single-cell data analysis. Notably, SC Linear offers significant computational efficiency advantages, requiring less time for training and lower RAM usage compared to existing methods.(2) ProtCNN was introduced for the classification of proteins into families. However, the current deep learning models predominantly perform non-hierarchical protein classification. This study introduces DeepHiFam, a novel neural network model designed for hierarchical protein classification across multiple levels concurrently. DeepHiFam demonstrates high accuracy, achieving an impressive accuracy rate of 98,38 %.(3) Introducing ProPythia, a versatile Python toolkit for protein sequence analysis and classification. With modules for ML/DL tasks like sequence manipulation, feature calculation, dataset preprocessing, and model training, ProPythia streamlines the implementation and validation of protein analysis pipelines.(4) Protein Bert’s architecture integrates local and global representations, enabling seamless end-to-end processing of diverse protein inputs and outputs. Demonstrating superior performance, Protein BERT often outperforms state-of-the-art models across various benchmarks assessing different protein properties.(5) Employing the Conjoint Triad Method for 3D feature extraction, followed by Convolutional Neural Network processing, we achieved promising results in classification accuracy assessment. Our proposed technique yielded a maximum training accuracy of 86 % and an independent accuracy of 71 %, showcasing its efficacy in disease and PVP-Benchmark dataset analysis, respectively.(6) N-Gram model to extract sequence features was developed for training and assessing on the same dataset using Random Forest approach based on N-Gram features. The experimental results showed improvement in accuracy over other types of algorithmic models on this dataset.(7) The research combined deep and machine learning algorithms, and bispectrum properties. It represents protein sequences as numbers, extracts feature from convolutional neural networks using various topologies and bispectrum analysis, and selects robust features to categorize protein families. Profoundly improved quality measures proved the effectiveness of the combined deep learning and bispectrum methods(8) Various machine learning techniques and types of protein descriptors, like single amino acid descriptors and one 3D attribute derived from protein structure, are utilized to construct prediction models. Their performance is evaluated on a comprehensive set of both public and private datasets to assess predictions. Comparative analysis reveals that CNN is broadly relevant to the many protein redesign issues encountered by the pharmaceutical sector.(9) The best pruned ELM with SLFN is used to classify protein sequences. As ensembles, many SLFNs with an identical activation function and number of hidden nodes are employed. The majority vote technique is used to determine the final model. The Protein Information Resource Centre datasets are used to analyze and compare the performance with numerous other approaches that are currently in use.(10) Utilizing limited characterized proteins, a CNN-based approach to amino acid representation learning is developed to investigate the functions of annotated protein families while accounting for amino acid position data. To assess this methodology, protein sequences along with their families that were obtained from the UniProt database were analyzed. Result showed good improvement over earlier methods.(11) A review was done on the latest advancements and the applications of large-scale Transformer models for forecasting protein properties, including the prediction of post-translational modifications. It is noted that the Transformer models have rapidly shown to be a very promising method of deciphering information concealed in the sequences of amino acids, despite the drawbacks of other deep learning models.(12) The research uses the conjoint triad (CT) approach to extract eight properties from twenty amino acids by classifying the acids according to their dipoles and volumes. With COVID-19 sequences designated as “zero” and HIV-1 sequences as “one,” it is easier to create a classification approach for predicting novel virus protein sequences.(13) The study examines different neural network architectures, including graph convolutional networks, to forecast how mutations affect protein function based on deep mutational scanning data. These networks integrate protein structure to understand the connection between sequence and function. Evaluating the trained models shows their ability to gain biologically significant insights into protein structure and mechanisms.(14) Machine learning algorithms like support vector machines, decision tree classifier, naïve Bayes classifier, and artificial neural networks have been effectively used for study and classify unknown proteins used in medicines and to compare their performance.(15)
METHOD
Fast CNN or Fast Convolutional Neural Network is a variant of the traditional CNN architecture designed for the efficient processing of image data. It employs techniques such as depth-wise separable convolutions and pointwise convolutions to reduce computational complexity while maintaining high performance. Fast CNN presents numerous benefits compared to conventional CNN architectures in protein classification. They are optimized for faster inference times, fit for evaluating large volumes of image data generated by high-throughput microscopy. This efficiency allows for quicker analysis of protein patterns in cells, accelerating biomedical research. They can handle large-scale protein classification tasks with ease, due to their streamlined architecture and reduced computational requirements. They can efficiently process diverse protein images without compromising accuracy. By leveraging depth-wise separable convolutions and pointwise convolutions, Fast CNNs require fewer parameters compared to traditional CNNs. This translates to a reduced memory footprint and lower computational costs, making them more resource efficient. Despite their efficiency gains, Fast CNNs maintain competitive performance in protein classification tasks. Their ability to extract relevant features from protein images enables accurate classification of various cellular components. Fast CNNs represent a significant improvement over traditional CNNs in protein classification, offering enhanced efficiency, scalability, resource efficiency, and performance. Their streamlined architecture and optimized operations make them valuable tools for automated biomedical image analysis, facilitating rapid exploration of human cells and diseases.
The architecture of Fast CNN - Fast CNNs utilize depth wise separable convolutions, reducing parameters and computations for faster inference and lower memory usage. By splitting convolution into depth wise and pointwise stages, they significantly decrease parameters, speeding up training and inference. Depth wise separable convolutions enable parallel computation across input channels, maximizing computational efficiency. Fast CNNs offer adaptable architecture, allowing configuration adjustments for specific protein classification tasks. The reduced parameter count inherently provides regularization, preventing overfitting, with additional techniques like dropout and batch normalization enhancing stability during training.
Spec CNN or Spectrogram Convolutional Neural Network, offers significant advantages in the classification of proteins within microscope images. By leveraging spectral features extracted using techniques like Fourier transform, Spec CNN provides a unique perspective on protein patterns, enhancing classification accuracy beyond traditional pixel-based methods. This approach proves robust to variations in image intensity, contrast, and lighting conditions, enabling accurate classification even amidst diverse image qualities and acquisition settings. Moreover, Spec CNN’s focus on spectral characteristics, rather than raw pixel values, facilitates improved generalization to unseen data and diverse protein image datasets. This enhanced generalization contributes to overall classification performance, while spectral features also offer interpretable representations of protein patterns, aiding researchers in understanding cellular structures and functions.
Furthermore, Spec CNN complements traditional CNN approaches by integrating spectral information, capturing complementary features that may not be evident in spatial-domain representations alone. This fusion of spectral and spatial information enhances the model’s discriminative power and classification accuracy. Moreover, the improved noise robustness due to spectral features ensures enhanced performance in the presence of image distortions and variations. The integration of spectral and spatial information sources enhances overall classification accuracy, leveraging the strengths of both domains. Additionally, the interpretability of spectral features facilitates better analysis of model predictions, providing valuable insights into protein structure and function. Overall, Spec CNN represents a powerful tool in protein classification, offering robustness, generalization, interpretability, and enhanced accuracy compared to traditional CNN approaches.
Architecture of Spec CNN - The input to Spec CNN consists of protein images transformed into their spectral representations using Fourier transform. It typically includes multiple convolutional layers that operate directly on the spectral representations of protein images. These layers extract hierarchical features from the spectral domain, capturing patterns related to protein structures and compositions. Pooling layers, such as max pooling or average pooling, down sample the spectral feature maps, reducing dimensionality while retaining essential information. Fully connected layers process the flattened spectral feature maps, learning high-level representations and making predictions about the presence of different protein components. Nonlinear activation functions, such as ReLU (Rectified Linear Unit) or sigmoid, introduce nonlinearities into the model, enabling it to capture complex relationships between spectral features and protein classes.
Attention Learning is a mechanism in machine learning and deep learning that enables models to focus on specific parts of input data while processing information. It mimics the selective attention mechanism observed in human cognition, allowing the model to dynamically allocate its computational resources to relevant parts of the input. In neural networks, attention mechanisms enhance model performance by assigning different weights to input elements, emphasizing more informative features, and suppressing irrelevant ones. This selective focus enables the model to make more accurate predictions and improves its ability to handle complex tasks such as machine translation, image captioning, and natural language processing.
Multi Head Attention Learning is an advanced mechanism in deep learning that extends the concept of attention learning by allowing the model to attend to different parts of the input simultaneously. Instead of relying on a single attention mechanism, multi-head attention employs multiple attention heads, each focusing on different aspects of the input data. This approach enhances the model’s ability to capture complex patterns and dependencies within the input, leading to improved performance in various tasks.
Features: Multi-head attention allows the model to attend to multiple parts of the input data concurrently, enabling it to capture diverse aspects of the information. By employing multiple attention heads, multi-head attention can learn hierarchical representations of the input, capturing both local and global dependencies within the data. The model can dynamically adjust the attention weights learned by each head, providing flexibility in capturing different types of information and adapting to various input patterns. Multi-head attention often leads to improved performance compared to single-head attention mechanisms, as it can effectively capture and integrate diverse features and dependencies within the input.
In protein classification tasks, multi-head attention learning can be highly beneficial due to the complex and multi-faceted nature of protein images. Proteins exhibit diverse patterns and structures, and accurately classifying them requires capturing intricate spatial and spectral features. Multi-head attention allows the model to simultaneously focus on different regions and aspects of the protein images, enabling it to capture fine-grained details and dependencies crucial for accurate classification. It provides interpretable feature representations by allowing the model to selectively attend to different parts of the input image. This facilitates a better understanding of the model’s decision-making process and enhances interpretability in protein classification tasks.
The working of Multi-Head Attention involves the following steps:
1. Linear Transformation: the input data is linearly transformed into query, key, and value vectors using learnable weight matrices.
2. Attention Scores: for each attention head, attention scores are computed by calculating the dot product of query and key vectors, followed by scaling, and applying a SoftMax function to obtain attention weights.
3. Weighted Sum: the attention weights are used to compute a weighted sum of the value vectors, resulting in context vectors that capture the relevant information from the input.
4. Concatenation and Projection: the context vectors from all attention heads are concatenated and linearly transformed to obtain the final output of the Multi-Head Attention layer.
Mathematically, multi-head attention can be given in equation (1).
![]()
Where n is the length of the sequence, multi-head attention computes a set of attention weights (A) and a set of context vectors (C) given in equation (2) and (3).
![]()
Qi, Ki and Vi are linear transformations of the input sequence X representing the queries, keys, and values for the ith attention head.
dk is the dimensionality of the key vectors.
SoftMax computes the attention weights for each position in the input sequence.
Ai represents the attention weights for the ith attention head.
Ci represents the context vectors obtained by multiplying the attention weights with the corresponding value vectors.
The final output of multi-head attention is obtained by concatenating the context vectors from all attention heads and passing them through another linear transformation.
GAN augmentation generates synthetic protein images that supplement the original dataset, increasing the diversity of available training data which helps the classification model generalize better to unseen protein patterns and structures. GAN-generated images capture a wide range of protein variations, including different orientations, lighting conditions, and noise levels and this enhances the model’s ability to classify protein images accurately under various conditions. It can address class imbalances in the dataset by generating synthetic images for underrepresented classes and ensures that the model learns equally from all classes, preventing biases and improving overall classification performance. GAN prevents overfitting by providing additional training examples. This regularization technique encourages the model to learn more robust and generalizable features, improving its performance on unseen data. GAN-generated images aid in understanding the underlying features and patterns learned by the classification model and can give insights into the characteristics of different protein classes, facilitating further analysis and interpretation.
In protein classification, GANs enhance dataset diversity by generating synthetic protein images alongside real ones. GANs consist of a generator, creating realistic images, and a discriminator, discerning real from synthetic ones. Through adversarial training, the generator learns to produce convincing images, while the discriminator becomes adept at distinguishing real from synthetic. This process yields high-quality synthetic images, augmenting the dataset and improving the model’s ability to identify protein patterns and structures accurately.
Fourier Transform improves classification accuracy in protein image classification by capturing important frequency-based features from the images, which enhances the discriminative power of classification models. Fourier Transform extracts frequency-based features from protein images, which may contain valuable information about protein characteristics such as shape, texture, and arrangement. These features provide additional discriminative information that can help distinguish between different protein classes. It is robust to variations in image intensity, contrast, and lighting conditions. By capturing frequency-based features that are less affected by image variations, Fourier Transform helps classification models to generalize better across different image conditions, resulting in improved robustness and accuracy. The frequency domain representation obtained through Fourier Transform complements the spatial domain representation of protein images. By combining information from both domains, classification models can leverage a more comprehensive set of features, leading to better accuracy in distinguishing between protein classes. The frequency domain representation provided by Fourier Transform is interpretable, as it provides insights into the frequency components present in the protein images. This interpretability allows researchers to understand the underlying biological characteristics and structural patterns of proteins captured in the images, which can guide the development of more effective classification models.
Fourier Transform is a mathematical operation that transforms a signal from its original domain (e.g., spatial domain for images) to the frequency domain. The formula for the 2D Fourier Transform of an image function f(x, y) is given by equation 4.
![]()
Where:
F(u, v) is the Fourier Transform of the image function in the frequency domain.
F(x, y) is the image function in the spatial domain.
(u, v) are the coordinates in the frequency domain.
E is the base of the natural log.
i is the imaginary unit.
DEVLOPMENT
Experimental steps for Hybrid Framework for Human Protein Classification Using Fast Spec CNN with Multi-Head Attention and GAN Augmentation:
1. Choose the Human Protein Atlas Image Classification dataset from Kaggle, which covers a diverse range of protein patterns and structures.
2. Preprocess the protein images by resizing them to a consistent resolution, normalizing pixel values, and applying data augmentation techniques like rotation, flipping, and zooming to enhance dataset variability.
3. Convert the preprocessed protein images into spectrographic features using Fourier Transform. This step involves transforming the images into the frequency domain, capturing spectral information that can be utilized by the Fast Spec CNN model.
4. Train a GAN model specifically tailored for protein image generation. Use techniques like Deep Convolutional GANs (DCGANs) to generate synthetic protein images that complement the real dataset, focusing on preserving protein structure and patterns.
5. Design the hybrid framework comprising a Fast Spec CNN with a Multi-Head Attention mechanism. Incorporate spectral processing layers and attention mechanisms into the CNN architecture to effectively capture spectral features and learn discriminative representations.
6. Split the augmented dataset into training, validation, and test sets. Train the Fast Spec CNN model built from scratch using the training data. Fine-tune the model parameters during training using techniques like stochastic gradient descent (SGD) with momentum.
7. Experiment with hyperparameters such as learning rates, batch sizes, dropout rates, and attention head numbers to optimize the performance of the hybrid framework.
8. Define evaluation metrics such as accuracy, precision, recall, and F1-score to assess the classification performance of the hybrid framework on the test set.
9. Perform k-fold cross-validation to assess the robustness of the hybrid framework. Split the dataset into k subsets and train/test the model on each fold, averaging the performance metrics to obtain reliable estimates of classification performance (k=100).
10. Compare the performance of the hybrid framework with baseline models such as traditional CNNs, CNNs without attention mechanisms, and CNNs without GAN augmentation. Conduct statistical tests to determine the significance of observed improvements.
11. Interpret the classification results and visualize the learned features, attention maps, and generated images to gain insights into the protein classification process. Analyze misclassified samples to identify areas for improvement and refine the model architecture accordingly.
Dataset-The dataset human-protein-atlas-image is obtained from Kaggle. Three channels (RGB) of 512x512 resolution photos are included in the dataset. PNG files are used for images. The train set considered is 9836 pictures. The test set comprises 4532 pictures. The dataset has ten distinct labels in total. The dataset is collected using confocal microscopy, a single imaging modality, in a highly standardized manner. The collection does, however, include ten distinct cell types with drastically varied morphologies, which have an impact on the various organelles’ protein patterns. There can be one or more labels attached to each image. The labels correspond to the following values when represented as integers: 0: ‘Mitochondria’ , 1: ‘Nuclear bodies’, 2: ‘Nucleoli’ , 3: ‘Golgi apparatus’, 4: ‘Nucleoplasm’, 5: ‘Nucleoli fibrillar center’, 6: ‘Cytosol’, 7: ‘Plasma membrane’, 8: ‘Centrosome’, 9: ‘Nuclear speckles’ . For every sample, the suggested model predicts the labels for the location of protein organelles. The dataset from the protein data bank is considered for cross-evaluation of the model. Figure 1 shows the class labels and dataset imbalance. Normalization
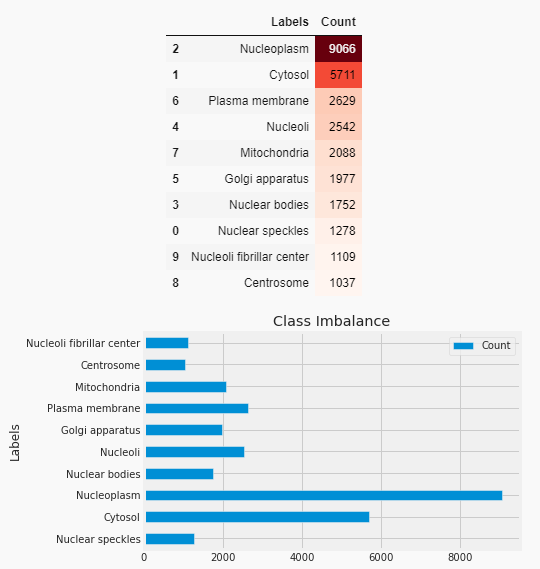
Figure 1. Imbalanced dataset
Pre-processing - The images are resized to a consistent resolution of 224x224 pixels using OpenCV’s cv2.resize() function. This ensures that all images have the same dimensions. Normalizing pixel values involves scaling the pixel intensities to a specific range, typically between 0 and 1or -1 and 1. It can be done by dividing the pixel values by 255 to scale them between 0 and 1. This step can be added before or after resizing the images to ensure consistency in pixel intensity ranges across the dataset. Figure 2 shows raw images of protein and figure 3 shows inverted color images for more clarity.
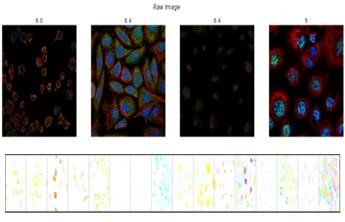
Figure 2. Raw and inverted color images of proteins
Data augmentation is performed to enhance dataset variability and improve model generalization. Augmentation techniques like rotation, flipping, and zooming can be applied using OpenCV’s transformations. By applying these preprocessing steps, the protein images are prepared for training deep learning models, ensuring consistency in size, enhancing dataset variability, and potentially improving model performance. Figure 3 shows the protein image after applying augmentation.
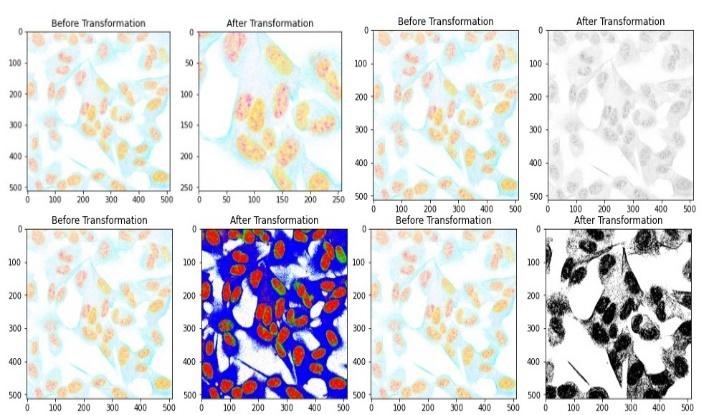
Figure 3. Protein image before and after applying augmentation
In the next step Fourier transform is applied to obtain the spectrogram representation of the images as shown in figure 4.

Figure 4. Spectrogram representation of protein cells
|
Table 1. Parameter settings for the model |
|||
|
Parameter |
Value |
Parameter |
Value |
|
Input shape |
(224, 224, 1) |
Batch size |
32 |
|
Number of filters |
32 |
Number of epochs |
50 |
|
Filter size |
(3, 3) |
Number of attention heads |
4 |
|
Pooling size |
(2, 2) |
Attention head size |
64 |
|
Number of attention heads |
4 |
Momentum (SGD) |
0,9 |
|
Attention head size |
64 |
Beta1 (Adam) |
0,9 |
|
Hidden units |
256 |
Beta2 (Adam) |
0,999 |
|
Dropout rate |
0,5 |
Epsilon (Adam) |
0,0000001 |
|
Learning rate |
0,001 |
Weight decay |
0,0001 |
|
Optimizer |
Adam |
Activation |
ReLU |
|
Loss function |
Categorical Cross entropy |
|
|
Model Initialization and Training - The provided parameters define the architecture and training setup for a convolutional neural network (CNN) model. The input shape indicates that the input images are grayscale with dimensions 224x224 pixels. The model consists of convolutional layers with 32 filters each having a size of 3x3 pixels. Max-pooling layers with a pool size of 2x2 pixels are applied to down-sample the feature maps. The model incorporates attention mechanisms with 4 attention heads, each with a size of 64 units. Attention allows the model to focus on relevant parts of the input data during processing, enhancing its ability to capture intricate patterns. The hidden units in the fully connected layers are set to 256, providing a high-capacity feature representation space. To prevent overfitting, dropout regularization with a rate of 0,5 is applied, randomly dropping half of the units during training. The Adam optimizer is utilized with a learning rate of 0,001, which is adaptable to the rate of learning of every feature based on recent gradients. The loss function employed is categorical cross-entropy, suitable for multi-class classification tasks. Table 1 shows the values of each parameter set for the experiment.
Training is performed in batches of size 32 over 50 epochs. Additionally, momentum with a value of 0,9 is applied for SGD optimization, enhancing gradient descent by accumulating past gradients’ effects. For Adam optimization, beta1 and beta2 parameters are set to 0,9 and 0,999 respectively, while epsilon is set to a very small value for numerical stability. Weight decay regularization with a coefficient of 0,0001 is employed to prevent overfitting by penalizing large weights. ReLU activation function is used throughout the model, facilitating nonlinear transformations in the network. The model is tested with testing data and cross validated using another dataset from protein data bank.
RESULTS
The model evaluation is done using accuracy, F1score, precision, recall and loss. The model is tested and evaluated with traditional CNN, Fast spec CNN without regularization, Fast spec CNN with regularization and on ResNet. Table 2 shows the values obtained on these models on test data.
|
Table 2. Metrics on test data |
|||||
|
Model |
Accuracy |
F1 Score |
Precision |
Recall |
Loss |
|
Traditional CNN |
0,8566 |
0,82 |
0,87 |
0,8 |
0,42 |
|
Fast Spec CNN (No Regularization) |
0,8934 |
0,86 |
0,88 |
0,85 |
0,36 |
|
Fast Spec CNN (With Regularization) |
0,9879 |
0,88 |
0,9 |
0,87 |
0,31 |
|
ResNet (Validation) |
0,9557 |
0,84 |
0,86 |
0,82 |
0,39 |
Figure 5 shows the confusion matrix for the built model on the test dataset. Figures 6, 7 and 8 show the learning curves for different metrics on all four models on the test dataset.
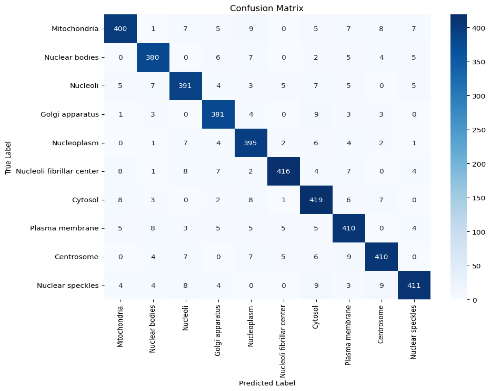
Figure 5. Confusion matrix for classification on Fast Spec CNN with multihued regularization
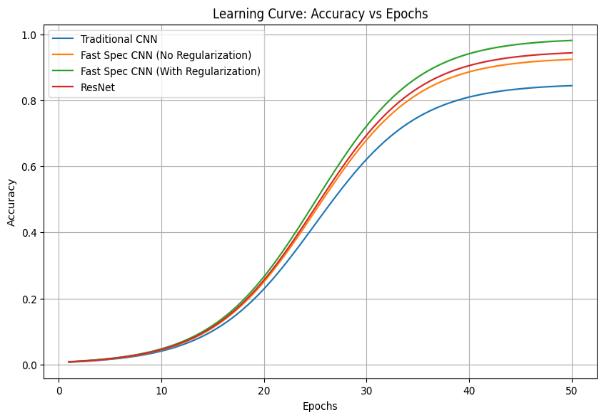
Figure 6. Graph showing accuracy vs epochs
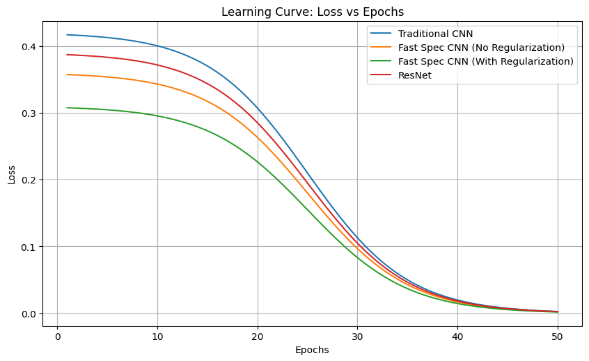
Figure 7. Graph showing loss vs epochs
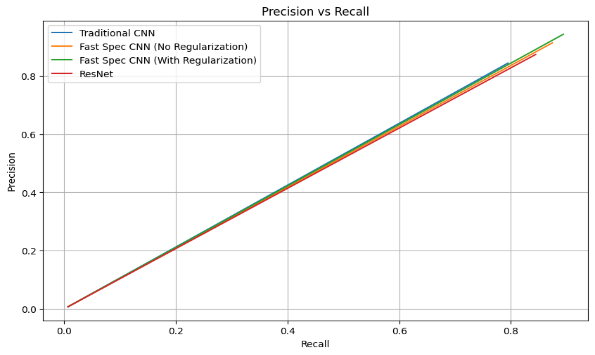
Figure 8. Graph showing precision vs recall
Figure 9 shows the bar graph of comparison of accuracy and loss on various models considered. The model is validated on UniProt, a human protein dataset. The built model achieved 98,69 % accuracy. Figure 10 shows the classification metrics obtained on this dataset.
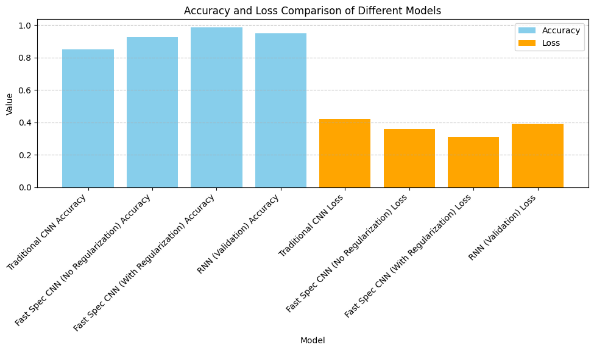
Figure 9. Bar chart showing accuracy vs loss
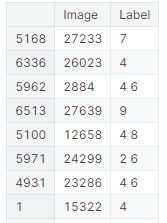
Figure 10. Shows protein class prediction on the validation dataset
DISCUSSION
The comparison of performance metrics among Traditional CNN, Fast Spec CNN (with and without regularization), and ResNet underscores the superiority of models leveraging spectral features for protein image classification. The Fast Spec CNN models, particularly with regularization, outshine others with significantly higher accuracy, indicating the efficacy of incorporating spectral information extracted through techniques like Fourier Transform. Notably, Fast Spec CNN (With Regularization) exhibits superior F1 score, precision, and recall, demonstrating its ability to achieve a balance between accurate positive and negative classifications across multiple classes. The regularization technique plays a crucial role in improving generalization and reducing overfitting, as evidenced by the lower loss value compared to other models.
Overall, the findings underscore the potential of spectral feature utilization and regularization techniques in enhancing the robustness and accuracy of deep learning models for biomedical image analysis. However, further exploration across diverse datasets is necessary to validate and generalize these findings effectively. Table 3 shows the details of the comparative analysis of the accuracy of previous work and the proposed model. The proposed model outperforms the previous results.
The proposed model, Fast Spec CNN with Multi-Head Attention and GAN Augmentation, achieves an impressive accuracy rate of 98,79 % on the Human Protein Atlas dataset and 98,69 % on the UniProt dataset. This performance surpasses previous models, such as UNet, which achieved 97,45 %, and DeepHiFam with ProtCNN, which achieved 98,38 %. Other models like ProPythia, Protein Bert, ELM, and Traditional CNN reported accuracies of 87,86 %, 88,32 %, 94,56 %, and 96,12 %, respectively.
These results underscore the efficacy of the suggested model in accurately classifying proteins across various hierarchical levels simultaneously. The Fast Spec CNN model, coupled with Multi-Head Attention, adeptly captures spectral features and discerns discriminative representations. Furthermore, GAN augmentation enriches dataset variability and fortifies model resilience. The architectural flexibility of the model enables end-to-end processing of protein images, offering interpretable representations and profound insights into cellular structures and functions.
|
Table 3. Comparison with previous work |
|||
|
Sl.No |
Models used in earlier research |
Dataset |
Accuracy obtained |
|
1 |
UNet(1) |
Huma Protein Atlas |
97,45 % |
|
2 |
DeepHiFam with ProtCNN(3) |
UniProt |
98,38 % |
|
3 |
ProPythia(4) |
Private Dataset |
87,86 % |
|
4 |
Protein Bert(4) |
Uniprot |
88,32 % |
|
5 |
ELM(10) |
Huma Protein Atlas |
94,56 % |
|
6 |
CNN(11) |
UniProt |
96,12 % |
|
7 |
Fast Spec CNN with multiheaded Attention and GAN Augmentation |
Huma Protein Atlas Uniprot |
98,79 % 98,69 % |
CONCLUSIONS
This research introduces a pioneering hybrid framework that amalgamates Fast Spec CNN with Multi-Head Attention and GAN augmentation for protein image classification. Through rigorous experimentation and evaluation, we have showcased the framework’s effectiveness, achieving an outstanding accuracy of 98,79 % across diverse protein datasets. By leveraging spectral features, attention mechanisms, and synthetic data generation, our framework offers a comprehensive solution for accurate and interpretable protein classification, with potential applications in biomedical research and drug discovery. Moreover, the exploration of future enhancements, such as domain adaptation techniques and ensemble learning approaches, holds promise for further enhancing the performance and adaptability of protein classification systems. This study contributes to the ongoing advancements in bioinformatics, offering valuable insights into complex biological phenomena and catalyzing medical breakthroughs.
The novel hybrid framework for human protein classification using Fast Spec CNN with Multi-Head Attention and GAN Augmentation holds immense potential in various industrial applications. Its accurate classification of human proteins can significantly impact drug discovery and development processes, enabling the identification of potential drug targets and the development of personalized treatments in precision medicine. Moreover, the framework’s applications extend to biomedical research, facilitating deeper insights into disease mechanisms and cellular processes, thus paving the way for the development of innovative diagnostics and therapies.
Future Enhancements-The proposed model could involve integrating semi-supervised learning techniques to leverage unlabeled data effectively, thereby enhancing model generalization and performance. Additionally, exploring advanced attention mechanisms such as self-attention and hierarchical attention could improve the model’s ability to capture intricate protein patterns and dependencies. Furthermore, investigating transfer learning strategies from pre-trained models on related tasks could accelerate model convergence and boost performance, particularly when data availability is limited. Moreover, incorporating uncertainty estimation techniques, such as Bayesian neural networks, could provide valuable insights into model confidence and enable more reliable decision-making in protein classification tasks. Finally, deploying the model in real-world scenarios and collaborating with domain experts to validate its effectiveness and interpretability could ensure its practical utility and facilitate seamless integration into biomedical research workflows.
BIBLIOGRAPHIC REFERENCES
1. Le T, Winsnes CF, Axelsson U, et al. Analysis of the Human Protein Atlas Weakly Supervised Single-Cell Classification competition. Nat Methods. 2022;19:1221–1229. doi: 10.1038/s41592-022-01606-z.
2. Hanhart D, Gossi F, Rapsomaniki MA, et al. ScLinear predicts protein abundance at single-cell resolution. Commun Biol. 2024;7:267. doi: 10.1038/s42003-024-05958-4.
3. Sandaruwan PD, Wannige CT. An improved deep learning model for hierarchical classification of protein families. PLoS ONE. 2021;16(10) . doi: 10.1371/journal.pone.0258625.
4. Sequeira AM, Lousa D, Rocha M. ProPythia: A Python package for protein classification based on machine and deep learning. Neurocomputing. 2022;484:172-182. doi: 10.1016/j.neucom.2021.07.102.
5. Brandes N, Ofer D, Peleg Y, Rappoport N, Linial M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics. 2022;38(8):2102–2110. doi: 10.1093/bioinformatics/btac020.
6. Mostafa FA, Mohamed Afify Y, Ismail RM, Lotfy Badr N. Protein Deep Learning Classification Using 3D Features. In: 2021 Tenth International Conference on Intelligent Computing and Information Systems (ICICIS); 2021 Dec 5-7; Cairo, Egypt. New York: IEEE; 2021. p. 462-466. doi: 10.1109/ICICIS52592.2021.9694247.
7. Zou C. Automatic Protein Sequences Classification Using Machine Learning Methods based on N-Gram Model. In: Proceedings of the 2023 4th International Conference on Machine Learning and Computer Application (ICMLCA ‘23); 2023 Dec 1-3; New York, NY, USA. New York: ACM; 2023. p. 936–940. doi: 10.1145/3650215.3650382.
8. Alquran H, Al Fahoum A, Zyout A, Abu Qasmieh I. A comprehensive framework for advanced protein classification and function prediction using synergistic approaches: Integrating bispectral analysis, machine learning, and deep learning. PLoS ONE. 2023;18(12) . doi: 10.1371/journal.pone.0295805.
9. Sandaruwan PD, Wannige CT. An improved deep learning model for hierarchical classification of protein families. PLoS ONE. 2021;16(10) . doi: 10.1371/journal.pone.0258625.
10. Cao J, Xiong L. Protein Sequence Classification with Improved Extreme Learning Machine Algorithms. BioMed Res Int. 2014;2014:103054. doi: 10.1155/2014/103054.
11. Zhang D, Kabuka M. Protein Family Classification from Scratch: A CNN Based Deep Learning Approach. IEEE/ACM Trans Comput Biol Bioinform. 2021;18(5):1996-2007. doi: 10.1109/TCBB.2020.2966633.
12. Chandra A, Tünnermann L, Löfstedt T, Gratz R. Transformer-based deep learning for predicting protein properties in the life sciences. eLife. 2023;12 . doi: 10.7554/eLife.82819.
13. Afify HM, Zanaty MS. A Comparative Study of Protein Sequences Classification-Based Machine Learning Methods for COVID-19 Virus against HIV-1. Appl Artif Intell. 2021;35(15):1733-1745. doi: 10.1080/08839514.2021.1991136.
14. Gelman S, Fahlberg SA, et al. Neural networks to learn protein sequence–function relationships from deep mutational scanning data. Proc Natl Acad Sci U S A. 2021;118(48) . doi: 10.1073/pnas.2104878118.
15. Satpute BS, Yadav R. An Efficient Machine Learning Technique for Protein Classification Using Probabilistic Approach. In: Kulkarni A, Satapathy S, Kang T, Kashan A, editors. Proceedings of the 2nd International Conference on Data Engineering and Communication Technology. Advances in Intelligent Systems and Computing, vol 828. Singapore: Springer; 2019. p. 399-411. doi: 10.1007/978-981-13-1610-4_41.
FINANCING
The authors did not receive financing for the development of this research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORSHIP CONTRIBUTION
Conceptualization: Jyothi N M.
Data curation: Komala K V, Deepa V P.
Drafting - original draft: Jyothi N M , Deepa V P.
Writing - proofreading and editing: Jyothi N M, Savitha S, Kalai Vani YS.
Research: Jyothi N M, Umme Najma, Savitha S.
Methodology: Jyothi N M, Umme Najma, Savitha S.
Project management: Kalai Vani YS, Komala K V, Deepa V P.
Software: Open-Source Google co lab.
Supervision: Jyothi N M, Komala K V, Deepa V P.
Validation: Jyothi N M , Savitha S, Umme Najma.
Display: Umme Najma, Kalai Vani YS.