Categoría: Health Sciences and Medicine
REVISIÓN BIBLIOGRÁFICA
Interpretation of fatty acids and adrenal function in Ald-X patients using PRISMA 2020 methodology
Interpretación de ácidos grasos y función suprarrenal en pacientes con Ald-X mediante metodología PRISMA 2020
Olivia Elizabeth Altamirano Guerrero1 ![]() *, Piedad Elizabeth Acurio Padilla1
*, Piedad Elizabeth Acurio Padilla1 ![]() *, Mauricio Fernando
Enrríquez Grijalva1
*, Mauricio Fernando
Enrríquez Grijalva1 ![]() *, Melany Yamilex
Reascos Chalacán1
*, Melany Yamilex
Reascos Chalacán1 ![]() *
*
1Universidad Regional Autónoma de Los Andes, Matriz Ambato, Ecuador.
Citar como: Altamirano Guerrero OE, Acurio Padilla PE, Enrríquez Grijalva MF, Reascos Chalacán MY. Interpretation of fatty acids and adrenal function in Ald-X patients using PRISMA 2020 methodology. Salud, Ciencia y Tecnología - Serie de Conferencias. 2023; 2:753. https://doi.org/10.56294/sctconf2023753
Enviado: 17-06-2023 Revisado: 25-09-2023 Aceptado: 19-12-2023 Publicado: 20-12-2023
Editor: Dr.
William Castillo-González ![]()
ABSTRACT
X-linked Adrenoleukodystrophy is a hereditary metabolic disorder that causes the accumulation of very long-chain fatty acids in various tissues, primarily affecting the nervous system and adrenal glands. The aim of the study was to interpret very long-chain fatty acids and adrenal function in patients with X-linked Adrenoleukodystrophy using PRISMA 2020 methodology. A systematic literature review and an analysis of a specific clinical case were conducted. A total of 135 records were reviewed, of which 16 relevant articles were selected after applying inclusion and exclusion criteria. The results highlight the complex relationship between VLCFA levels and adrenal function, and the importance of an integrated approach to treatment. It is concluded that further research is essential to better understand this correlation and develop effective therapeutic strategies.
Keywords: Adrenoleukodystrophy; X Chromosome; Fatty Acids; Adrenal Insufficiency; Addison’s Disease.
RESUMEN
La Adrenoleucodistrofia ligada al cromosoma X es una enfermedad metabólica hereditaria que provoca la acumulación de ácidos grasos de cadena muy larga en diversos tejidos, afectando principalmente el sistema nervioso y las glándulas suprarrenales. El objetivo del estudio fue interpretar los ácidos grasos de cadena muy larga y la función suprarrenal en pacientes con Adrenoleucodistrofia ligada al cromosoma X mediante metodología PRISMA 2020. Se realizó una revisión sistemática de la literatura y un análisis de un caso clínico específico. Se revisaron 135 registros, de los cuales se seleccionaron 16 artículos relevantes tras aplicar criterios de inclusión y exclusión. Los resultados destacan la compleja relación entre los niveles de AGCML y la función suprarrenal, y la importancia de un enfoque integral en el tratamiento. Se concluye que es fundamental una mayor investigación para comprender mejor esta correlación y desarrollar estrategias terapéuticas efectivas.
Palabras clave: Adrenoleucodistrofia; Cromosoma X; Ácidos Grasos; Insuficiencia Suprarrenal; Enfermedad de Addison.
INTRODUCCIÓN
La Adrenoleucodistrofia ligada al X (ALD-X) es la patología peroxisomal más frecuente. Este trastorno metabólico se caracteriza por una alteración peroxisomal que provoca la acumulación anormal de ácidos grasos saturados de cadena muy larga (AGSCML) en el plasma y los tejidos. La incidencia de esta enfermedad es aproximadamente de 1 caso por cada 17 000-20 000 varones recién nacidos y puede ser responsable de hasta el 10 % de los casos de insuficiencia suprarrenal (ISR) en varones. El espectro clínico varía desde una forma cerebral progresiva que conduce a una discapacidad grave en la primera década de vida hasta una adrenoleucomielopatía que se inicia en la edad adulta o incluso una presentación de enfermedad de Addison (Posada Bustos, Charry Lopez, y Espinosa García 2021).
La anomalía bioquímica subyacente radica en una disfunción en el proceso de beta oxidación peroxisomal de los ácidos grasos saturados de cadena muy larga (AGSCML) con más de 22 átomos de carbono. La incapacidad para metabolizar estos AGSCML propicia una acumulación anormal a nivel tisular, lo que presenta múltiples manifestaciones diversas y a menudo progresivas que no pueden preverse al nacer. En el sistema nervioso central, al integrarse a los lípidos complejos que forman la mielina, se inicia un proceso de desestabilización con desmielinización y degeneración, lo que se cree involucra reacciones inmunológicas. En la corteza suprarrenal, los AGSCML son mal sustrato para hidrolasas y su acúmulo lleva a disfunción y muerte celular, con la consiguiente disminución de la producción de esteroides activos. La insuficiencia suprarrenal primaria es un fenotipo clínico importante en la ALD, con una prevalencia estimada de por vida de más del 80 % (Zhu et al. 2020).
Varios estudios evidencian que los defectos en el gen ABCD1 no son suficientes para explicar completamente la base molecular que subyace a la variabilidad fenotípica de la adrenoleucodistrofia. Hasta el momento, la investigación se ha centrado principalmente en la fisiopatología de la adrenoleucodistrofia a través de los ácidos grasos de cadena muy larga (VLCFA), incluyendo tanto los monoinsaturados como los saturados, y sus correspondientes vías bioquímicas. Se han publicado pocos estudios sobre los ésteres de colesterilo y los fosfolípidos, pero estos también se han limitado a abordar la acumulación de los ácidos de cadena muy larga (Cueva y Tinitana 2023).
Es relevante destacar que la relación entre los ácidos grasos de cadena muy larga saturados y los monoinsaturados como un factor contribuyente requiere un análisis más exhaustivo. Los estudios a nivel celular demuestran que la exposición de los fibroblastos ALD a los VLCFA saturados (C26:0) provoca estrés en el retículo endoplasmático y, por consiguiente, induce la muerte celular inducida por lípidos. Por otro lado, la exposición a los monoinsaturados (C26:1) no produce este resultado (Torres y Grijalva 2023).
El análisis más comúnmente empleado para el diagnóstico es la evaluación del perfil anormal de ácidos grasos saturados de cadena muy larga (AGSCML) en la sangre. La acumulación de estos AGSCML en el área del fascículo y a nivel cerebral posibilita la detección de esta afección (Ruiz-Sánchez et al. 2022).
La Adrenoleucodistrofia (ALD) y el hipoaldosteronismo comparten una conexión a través de su impacto en la función suprarrenal y el metabolismo de los ácidos grasos. En la ALD, una condición hereditaria que afecta la descomposición de los ácidos grasos de cadena muy larga, se produce una acumulación de estos ácidos en diversos tejidos, incluyendo las glándulas suprarrenales. En casos avanzados de ALD, se puede desarrollar una insuficiencia suprarrenal primaria, también conocida como enfermedad de Addison, en la cual las glándulas suprarrenales no producen suficientes hormonas, incluida la aldosterona.
Esta relación entre la ALD y la insuficiencia suprarrenal resalta cómo la acumulación de ácidos grasos puede impactar directamente en la función hormonal. Por otro lado, el hipoaldosteronismo, que se caracteriza por un déficit en la acción de la aldosterona, puede resultar en desequilibrios electrolíticos y problemas de equilibrio ácido-base.
En casos de ALD, donde la acumulación de ácidos grasos afecta las glándulas suprarrenales, se puede manifestar una insuficiencia suprarrenal, lo que significa que la aldosterona y otras hormonas no se producen en cantidades adecuadas. Esto, a su vez, puede tener implicaciones en la regulación del equilibrio de electrolitos y el balance ácido-base en el cuerpo, lo cual es una característica clave del hipoaldosteronismo (Morigaki 2022).
El objetivo del estudio es interpretar los ácidos grasos de cadena muy larga y la función suprarrenal en pacientes con Adrenoleucodistrofia ligada al cromosoma X mediante metodología PRISMA 2020.
MÉTODO
En este estudio se llevó a cabo una revisión bibliográfica sistemática, descriptiva, siguiendo la metodología PRISMA 2020 para interpretar los ácidos grasos de cadena muy larga y la función suprarrenal en pacientes con Adrenoleucodistrofia ligada al cromosoma X (ALD-X). La revisión se realizó en los idiomas inglés, portugués y español, abarcando artículos científicos relevantes en la comunidad médica.
PRESENTACIÓN DEL CASO
A continuación, se presenta un caso clínico que ilustra los hallazgos revisados. Este caso se integra en el estudio para proporcionar un ejemplo práctico y reciente de la aplicación de los conocimientos obtenidos a partir de la revisión bibliográfica.
Se presenta el caso de un paciente masculino de 2 años y 9 meses, producto de la tercera gesta de padres no consanguíneos. Nació a las 39,2 semanas gestacionales por fecha de última menstruación (FUM) y a las 41,1 semanas por escala Capurro, con un puntaje APGAR de 6 al minuto y de 8 a los cinco minutos. El peso al nacer fue de 2730 gramos, talla de 44 cm y perímetro cefálico de 36,5 cm. El parto fue cefalo-vaginal sin complicaciones.
El paciente acudió a consulta externa en el centro de salud tipo C del cantón Quero debido a vómitos y diarrea. Además, se evidenciaron alteraciones en el tono muscular, déficit neurológico brusco, espasmos musculares y espasticidad en miembros superiores e inferiores. Los padres refirieron que el desarrollo y crecimiento del niño habían sido normales hasta el inicio abrupto de los síntomas. En el centro de salud se administraron soluciones salinas al 0,9 % y se derivó a pediatría.
El pediatra solicitó una tomografía axial computarizada (TAC) cerebral simple, que mostró características de enfermedad degenerativa desmielinizante. Veintiséis días después, una resonancia magnética (RM) de cerebro reveló áreas de desmielinización simétrica de la sustancia blanca peritrigonales, compatibles con Adrenoleucodistrofia ligada al cromosoma X. Se realizaron análisis hematológicos que confirmaron la presencia de ácidos grasos de cadena muy larga (AGCML), desnutrición y anemia. El tratamiento consistió en cuidados paliativos, incluyendo suplementos vitamínicos, hierro, calcio, AINES como ibuprofeno, e hidrocortisona para la insuficiencia adrenal.
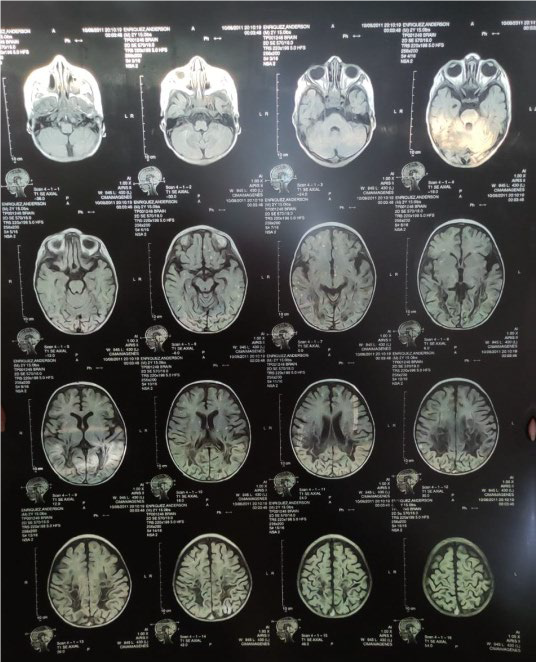
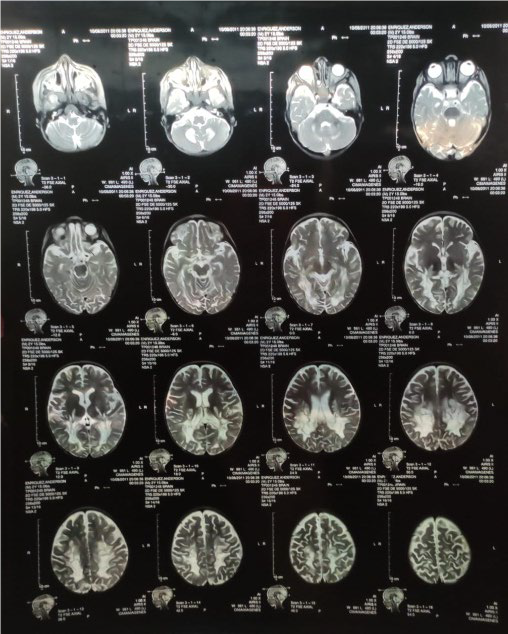
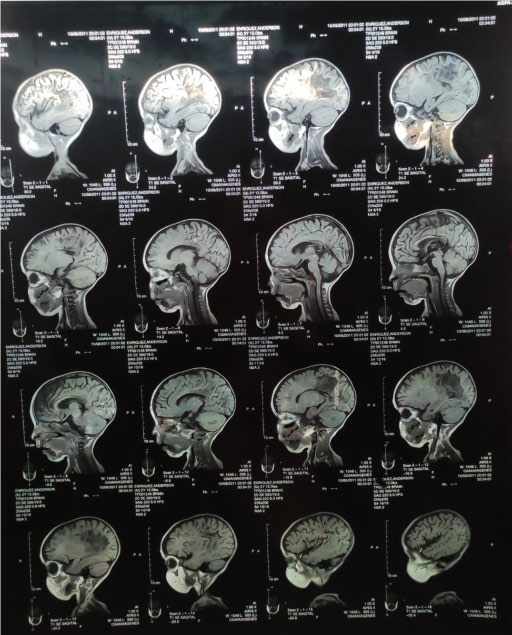
Figura 1. Cortes sagitales, axiales y coronales en secuencias T1, T2 y DP
En la figura 1 se observan los cortes sagitales, axiales y coronales en secuencias T1, T2 y DP. Adviértase áreas de desmielinización simétrica de sustancia blanca peri-trigonal que se extiende por el esplenio del cuerpo calloso, brazo posterior de las cápsulas internas, lóbulos caudales del cerebelo. (Resonancia magnética del paciente).
Por su parte, en la figura 2 se aprecia la fisiopatología de la acumulación de ácidos grasos de cadena muy larga desde el cuerpo al peroxisoma.
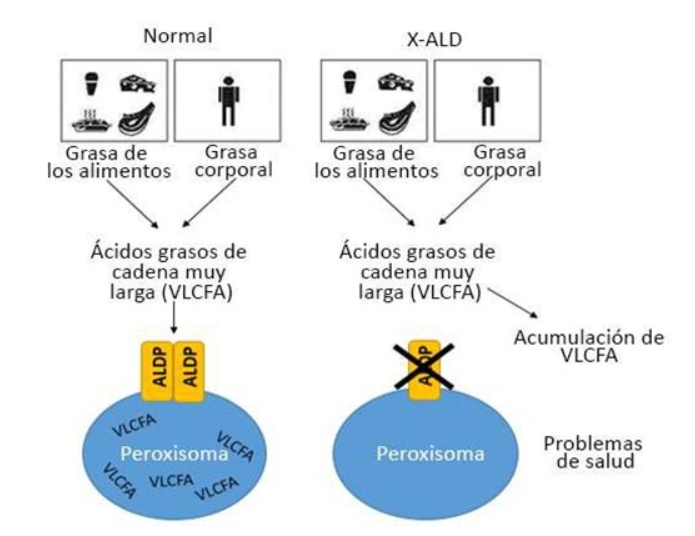
Figura 2. Fisiopatología de la acumulación de ácidos grasos
Revisión bibliográfica
Se utilizaron criterios de búsqueda específicos: “Insuficiencia adrenal”, “Adrenoleucodistrofia”, “Enfermedad de Addison”, y los operadores booleanos “OR” y “AND”. Las bases de datos consultadas incluyeron Scielo, Elsevier, PubMed, Biblioteca Virtual de la Salud y Google Académico, cubriendo el período de septiembre a octubre de 2023.
Criterios de inclusión y exclusión
Los criterios de inclusión abarcaron publicaciones desde el año 2019, artículos científicos en revistas indexadas y de alto impacto, revisiones bibliográficas sistemáticas, metaanálisis y referencias clásicas de la literatura médica que abordaran la ALD-X, la insuficiencia suprarrenal primaria y la enfermedad de Addison en relación con la acumulación de AGCML.
Proceso de selección
Se identificaron 135 registros mediante búsquedas en bases de datos y 5 registros adicionales de otras fuentes. Después de eliminar los duplicados, se contaron 109 registros. Se cribaron 50 registros y se evaluaron 59 artículos completos para su elegibilidad. Finalmente, se incluyeron 16 artículos en la síntesis cualitativa (Ver figura 3).
Este riguroso enfoque asegura una cobertura exhaustiva y actualizada de la literatura pertinente, facilitando una interpretación precisa de los ácidos grasos de cadena muy larga y la función suprarrenal en pacientes con ALD-X.
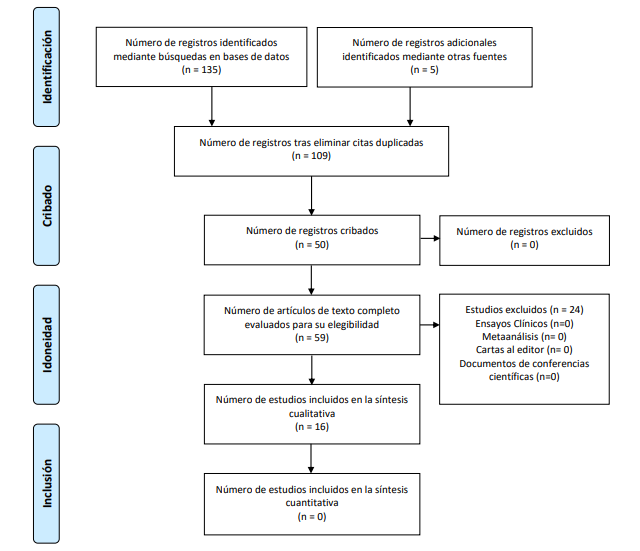
Figura 3. Diagrama PRISMA aplicado en la investigación
RESULTADOS
La adrenoleucodistrofia ligada al X (X-ALD) es un trastorno neuro metabólico letal caracterizado por desmielinización cerebral, degeneración axonal en la médula espinal, insuficiencia adrenal y acumulación de ácidos grasos de cadena muy larga (AGCML), principalmente C26:0. Un estudio basado en observaciones patológicas en la corteza suprarrenal, células de Schwann, testículos y cerebro postmortem, concluye que las inclusiones inusuales consisten en ésteres de colesterol con una cantidad excesiva de ácidos grasos de cadena muy larga (VLCFA) (Turk et al. 2020). Un número considerable de hombres afectados por X-ALD experimentan insuficiencia adrenocortical, que puede manifestarse después de la aparición de síntomas neurológicos o incluso décadas más tarde. En la mayoría de los casos, los hombres que presentan insuficiencia adrenocortical no muestran signos de desmielinización cerebral, clasificándose esto como “Addison solo” (Palakuzhiyil et al. 2020).
La enfermedad resulta de mutaciones en el gen ABCD1 que provocan la pérdida de función de la proteína ALD, la cual participa en el transporte de AGCML al interior del peroxisoma, donde se degradan por b-oxidación. Existen más de 2,707 mutaciones de ABCD1, de las cuales 812 son no recurrentes y 248 son variantes de significado desconocido (Datos sobre la ALD 2023). La expresión clínica de la enfermedad está determinada por la insuficiencia suprarrenal y la afectación neurológica (Morigaki 2022).
Si el trasplante alogénico de células madre hematopoyéticas (HSCT) se realiza tempranamente, antes de que aparezcan los síntomas neurológicos y antes de que la enfermedad cerebral alcance un estado avanzado, puede detener el proceso de desmielinización en el cerebro. La identificación precoz, a través de la detección de hombres en riesgo en sus familias y, a partir de diciembre de 2013 en los EE. UU., la detección de recién nacidos, ha generado optimismo en cuanto a la posibilidad de un tratamiento exitoso para la ALD (Turk et al. 2020). En este caso presentado, se sugirió el trasplante de células madre para evitar el deterioro del paciente, pero debido a la disponibilidad limitada de este procedimiento en el país y el estado avanzado del déficit neurológico, se optó por un tratamiento paliativo con medicamentos para calmar el dolor y fisioterapia para la espasticidad.
En las etapas iniciales de la enfermedad, se administraron suplementos vitamínicos como Complejo B y calcio para contrarrestar la anemia y promover el funcionamiento óptimo del sistema nervioso, evitando así la hipotonía, debilidad, pérdida de apetito e irritabilidad. Asimismo, se buscaba fortalecer la estructura ósea. Varios estudios sugieren una dieta baja en ácidos grasos para evitar su acumulación y prevenir una progresión más agresiva de la enfermedad (Burgos Peláez et al. 2010).
Actualmente, las actualizaciones en el tratamiento se enfocan en los siguientes ejes:
1. Importancia de la monitorización de los AGCML: La monitorización de los niveles de AGCML en pacientes con ALD-X es crucial para evaluar la progresión de la enfermedad y determinar el curso de tratamiento.
2. Impacto en la función suprarrenal: La acumulación de AGCML en las glándulas suprarrenales puede impactar significativamente la función suprarrenal, resultando en disfunciones hormonales, incluida la producción de aldosterona.
3. Enfoque de tratamiento integral: Un enfoque de tratamiento integral que incluya terapias para reducir la acumulación de AGCML y estrategias para abordar las disfunciones suprarrenales puede mejorar la calidad de vida de los pacientes.
4. Avances en terapias específicas: La investigación actual explora terapias dirigidas a la corrección de las alteraciones metabólicas causadas por la acumulación de AGCML, incluyendo enfoques farmacológicos y terapias génicas.
5. Necesidad de seguimiento a largo plazo: Dada la naturaleza progresiva de la ALD-X, es esencial un seguimiento a largo plazo para evaluar la evolución de la enfermedad y ajustar el tratamiento en consecuencia.
La anomalía bioquímica clave es la acumulación tisular de AGCML. El tratamiento se enfoca en la terapia sustitutiva con esteroides adrenales y en algunos casos, el uso de aceite de Lorenzo ha demostrado reducir los niveles de AGCML en pacientes con ALD (Adrenoleucodistrofia ligada al cromosoma 2023). El aceite de Lorenzo, una mezcla de glicerol trioleato/glicerol trierucato en una proporción 4:1, permite reducir los niveles de C26:0 al competir con los ácidos grasos saturados en el proceso de elongación (Adrenoleucodistrofia ligada al cromosoma 2023).
El tratamiento con esteroides adrenales se realiza en pacientes sintomáticos. El fármaco de elección es la hidrocortisona, que presenta un efecto dual, tanto glucocorticoide como mineralocorticoide (Reyes-Justiniano et al. 2021). Las mediciones periódicas de la función adrenal y la terapéutica sustitutiva con esteroides adrenales son esenciales para todos los enfermos. Este tratamiento no altera la progresión neurológica, pero previene la discapacitación y muerte debido a las crisis addisonianas (Mariscal-Delgadillo et al. 2023).
DISCUSIÓN
Numerosos análisis de pacientes con ALD-X revelan que en más del 85 % de los casos, los síntomas neurológicos preceden a los de insuficiencia adrenal (Palakuzhiyil et al. 2020). En niños varones de países desarrollados, las causas más comunes de insuficiencia suprarrenal primaria son de origen autoinmunitario y la adrenoleucodistrofia. Es crucial descartar estas causas antes de clasificar el caso como idiopático o atribuirlo a la enfermedad de Addison. Una causa menos frecuente de insuficiencia suprarrenal primaria es la infiltración suprarrenal por citomegalovirus, que generalmente se presenta en pacientes con compromiso de la inmunidad celular y provoca un aumento en el tamaño de las glándulas suprarrenales, evidente en las imágenes de tomografía computarizada (Tornero Patricio et al. 2009).
El espectro clínico de la ALD-X se caracteriza por una disfunción neurológica progresiva y una insuficiencia suprarrenal con un pronóstico devastador. Es fundamental realizar pruebas para determinar una etiología autoinmune y adrenoleucodistrofia en niños con insuficiencia suprarrenal primaria antes de diagnosticar la enfermedad de Addison. La enfermedad de Addison se caracteriza por un déficit en la producción de todas las hormonas esteroidogénicas, incluyendo el déficit de mineralocorticoides, glucocorticoides y andrógenos, diferenciándose de la insuficiencia suprarrenal secundaria, en la que el déficit es de glucocorticoides y andrógenos debido a la falta de estimulación por la hormona adrenocorticótropa (ACTH) (Fernández-Rodríguez et al. 2016).
La implicación del sistema nervioso, cerebro y médula espinal, así como las glándulas suprarrenales, es crucial en la traducción clínica de esta enfermedad. Hasta la fecha, se han identificado más de 340 mutaciones en el gen ABCD1 (Datos sobre la ALD 2023). Los niveles de ácidos grasos saturados de cadena muy larga se obtienen mediante cromatografía de gas en el plasma y/o fibroblastos de la piel después de la cultura. El diagnóstico bioquímico se basa en un aumento significativo en los niveles de ácido hexacosanoico (C26:0), en las relaciones ácido hexacosanoico/ácido docosanoico (C26:0/C22:0) y ácido tetracosanoico/ácido docosanoico (C24:0/C22:0) (Datos sobre la ALD 2023). En una investigación que siguió a un grupo de niños sin síntomas neurológicos aparentes, se observó que el 80 % presentaba indicios bioquímicos de insuficiencia adrenal (Morigaki 2022).
El empleo de la metodología PRISMA 2020 en este estudio resalta por su rigurosidad y estructura sistemática en la revisión de la literatura existente sobre la ALD-X. PRISMA 2020 proporciona un marco metodológico estandarizado que facilita la identificación, selección, evaluación y síntesis de la evidencia relevante, garantizando la transparencia y reproducibilidad del proceso de revisión. Este enfoque meticuloso ha sido clave para sintetizar los hallazgos sobre la correlación entre los niveles de ácidos grasos de cadena muy larga (AGCML) y la función suprarrenal en pacientes con ALD-X, destacando la importancia de un tratamiento integral.
En Ecuador, otros estudios recientes también evidencian el potencial de la metodología PRISMA 2020 en diversas áreas médicas. Por ejemplo, Sánchez Sandoval et al. (2024) utilizan PRISMA 2020 para interpretar la entrega de malas noticias en la práctica médica (Sánchez Sandoval, Reyes Espinoza, and Burbano Pijal 2024), mientras que Muñoz Padilla et al. (2024) aplican esta metodología para revisar el uso del hidróxido de calcio como medicamento intraductal (Muñoz Padilla, Vega Martínez, and Villafuerte Moya 2024). Asimismo, Torres Yánez et al. (2024) analizan las complicaciones quirúrgicas laparoscópicas de quistes ováricos mediante una revisión con PRISMA 2020 (Torres Yánez, Analuiza Rea, and Cevallos Fuel 2024). Estos estudios demuestran la versatilidad y eficacia de PRISMA 2020 en la investigación médica.
CONCLUSIONES
Basándonos en el caso estudiado y en la revisión bibliográfica sobre la correlación entre los niveles de ácidos grasos de cadena muy larga (AGCML) y la función suprarrenal en pacientes con Adrenoleucodistrofia ligada al cromosoma X (ALD-X), podemos concluir lo siguiente:
La ALD-X, una enfermedad metabólica hereditaria, afecta significativamente la descomposición y metabolismo de los AGCML, resultando en su acumulación en diversos tejidos y órganos, incluyendo las glándulas suprarrenales. La interrelación entre los niveles de AGCML y la función suprarrenal en pacientes con ALD-X es compleja y multifacética. La acumulación de AGCML en las glándulas suprarrenales puede inducir disfunciones en la producción hormonal, manifestándose en una variedad de síntomas clínicos.
Es crucial señalar que la relación entre los niveles de AGCML y la función suprarrenal puede ser altamente variable entre individuos y no siempre sigue un patrón uniforme. Otros factores genéticos y ambientales también pueden influir en esta correlación. En vista de esta complejidad, resulta imperativo llevar a cabo investigaciones adicionales para lograr una comprensión más completa de la interacción entre los niveles de AGCML y la función suprarrenal en pacientes con ALD-X. Esto no solo profundizará en la fisiopatología de la enfermedad, sino que también podría tener importantes implicaciones para el desarrollo de estrategias de diagnóstico y tratamientos más efectivos.
REFERENCIAS BIBLIOGRÁFICAS
1. Adrenoleucodistrofia Ligada al Cromosoma. [Internet]. [Citado 23 de octubre de 2023]. Disponible en: https://www.newbornscreening.info/es/adrenoleucodistrofia-ligada-al-cromosoma/
2. Burgos Peláez, R., M. Guerrero Gual, G. Cárdenas, y H. Segurola. 2010. “Trastornos Congénitos del Metabolismo de los Lípidos: Adrenoleucodistrofia. Dieta Controlada en Ácidos Grasos.” En Dietoterapia, Nutrición Clínica y Metabolismo, 345-352. Madrid: Díaz de Santos. [Internet]. [Citado 23 de octubre de 2023]. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=7509300
3. Cueva, P., y J. G. L. Tinitana. 2023. “Adrenoleucodistrofia ligada al cromosoma X. Reporte de caso y revisión de la literatura.” Tesla Rev Científica 3(1): e139-e139.
4. Datos sobre la ALD. [Internet]. [Citado 23 de octubre de 2023]. Disponible en: https://adrenoleukodystrophy.info/clinica-y-diagnostico/datos-sobre-la-ald
5. Dorta-Contreras, A. J., C. González-Losada, y D. Wainshtok-Tomás. 2018. “Adrenomieloneuropatía, Fenotipo de la Adrenoleucodistrofia Ligada al Cromosoma X. Reporte de un Caso.” Rev Habanera Cienc Médicas 17(1):29-38.
6. Fernández-Rodríguez, E., I. Bernabeu, C. Guillín, y F. F. Casanueva. 2016. “Enfermedades de las glándulas adrenales. Insuficiencia suprarrenal primaria.” Med - Programa Form Médica Contin Acreditado 12(14):775-80.
7. Llerena Cepeda, M. de L., L. K. Sailema López, y G. A. Zúñiga Cárdenas. 2022. “Variantes de COVID-19 predominantes en Ecuador y sus síntomas asociados.” Universidad y Sociedad 14(S3):93-04.
8. Mariscal-Delgadillo, M. A., O. Crespo-Zambrano, Y. D. Maldonado, R. A. Tardio-Fiares, y T. F. Luna. s.f. “Tratamiento de la adrenoleucodistrofia ligada al cromosoma X.”
9. Morigaki, M. T. 2022. “Prevalência de Adrenoleucodistrofia em Crianças de 5 a 12 Anos. Prevalence of Adrenoleukodystrophy in Children from 5 to 12 Years: An Integrative Literature Review.” [Internet]. [Citado 23 de octubre de 2023]. Disponible en: http://repositorio.ufgd.edu.br/jspui/handle/prefix/5450
10. Muñoz Padilla, M. B., Vega Martínez, V. A., and Villafuerte Moya, C. A. 2024. “Interpretación Mediante Revisión Bibliográfica Del Uso Del Hidróxido De Calcio Como Medicamento Intraductal.” Salud, Ciencia y Tecnología 4: 924. Disponible en: https://revista.saludcyt.ar/ojs/index.php/sct/article/view/924
11. Palakuzhiyil, S. V., R. Christopher, y S. R. Chandra. 2020. “Deciphering the modifiers for phenotypic variability of X-linked adrenoleukodystrophy.” World J Biol Chem 11(3):99-111.
12. Posada Bustos, S., M. L. Charry Lopez, y E. Espinosa García. 2021. “Adrenoleucodistrofia ligada a X: Un caso de presentación aguda cerebral infantil.” Andes Pediatr 92(4):602-8.
13. Reyes-Justiniano, A., E. C. Beltrán-Luna, y M. A. Caballero-Chacón. 2021. “Enfermedad de Addison e insuficiencia adrenal aguda: Presentación de un caso y revisión de la literatura.” Cuad Hosp Clínicas 62(1):63-71.
14. Ruiz-Sánchez, J. G., A. L. Calle-Pascual, M. Á. Rubio-Herrera, M. P. De Miguel Novoa, E. Gómez-Hoyos, y I. Runkle. 2022. “Clinical manifestations and associated factors in acquired hypoaldosteronism in endocrinological practice.” Front Endocrinol 13:990148.
15. Sánchez Sandoval, P. A., Reyes Espinoza, L. K., and Burbano Pijal, D. C. 2024. “Interpretación De La Entrega De Malas Noticias En La Práctica Médica a Través De La Revisión De La Literatura PRISMA 2020.” Salud, Ciencia y Tecnología 4: 931. Disponible en: https://revista.saludcyt.ar/ojs/index.php/sct/article/view/931
16. Tornero Patricio, S., J. A. B. de la Vega, D. Nehme Álvarez, F. J. Gentil González, M. D. Lluch Fernández, y J. González Hachero. 2009. “Insuficiencia suprarrenal primaria como inicio de adrenoleucodistrofia en un varón de 4 años.” Endocrinol Nutr 56(1):40-2.
17. Torres, R. E. del C. O., y M. F. E. Grijalva. 2023. “Investigación de la Adrenoleucodistrofia ligada al Cromosoma X como parte del programa de postgrado.” Rev Conrado 19(92):146-54.
18. Torres Yánez, J. A., Analuiza Rea, E. N., and Cevallos Fuel, T. A. 2024. “Análisis Mediante Revisión Bibliográfica Con Metodología PRISMA 2020 De Las Complicaciones Quirúrgicas Laparoscópicas De Quistes Ováricos.” Salud, Ciencia y Tecnología 4: 936. Disponible en: https://revista.saludcyt.ar/ojs/index.php/sct/article/view/936
19. Turk, B. R., C. Theda, A. Fatemi, y A. B. Moser. 2020. “X‐linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies.” Int J Dev Neurosci 80(1):52-72.
20. Zhu, J., F. Eichler, A. Biffi, C. N. Duncan, D. A. Williams, y J. A. Majzoub. 2020. “The Changing Face of Adrenoleukodystrophy.” Endocr Rev 41(4):577-93.
FINANCIACIÓN
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses.
CONTRIBUCIÓN DE AUTORÍA
Conceptualización: Olivia Elizabeth Altamirano Guerrero, Piedad Elizabeth Acurio Padilla, Mauricio Fernando Enrríquez Grijalva, Melany Yamilex Reascos Chalacán.
Curación de datos: Olivia Elizabeth Altamirano Guerrero, Piedad Elizabeth Acurio Padilla, Mauricio Fernando Enrríquez Grijalva, Melany Yamilex Reascos Chalacán.
Investigación: Olivia Elizabeth Altamirano Guerrero, Piedad Elizabeth Acurio Padilla, Mauricio Fernando Enrríquez Grijalva, Melany Yamilex Reascos Chalacán.
Redacción – borrador original: Olivia Elizabeth Altamirano Guerrero, Piedad Elizabeth Acurio Padilla, Mauricio Fernando Enrríquez Grijalva, Melany Yamilex Reascos Chalacán.
Redacción – revisión y edición: Olivia Elizabeth Altamirano Guerrero, Piedad Elizabeth Acurio Padilla, Mauricio Fernando Enrríquez Grijalva, Melany Yamilex Reascos Chalacán.